aminoXpress是一款行业必备的生物分析软件。一款功能强大的生物分析工具aminoXpress。它可以帮助用户在软件上分析生物学数据,支持氨基酸分析,化合物分析,元素分析,片段化,同位素谱,分子量,数据库管理,序列等功能。用户可以直接启动软件中的氨基酸分析模块。该模块有两个。该部分用于数据输入。对于某些氨基酸,例如酪氨酸,由于在水解过程中容易被氧化的特性,结果常常是不准确的。在未优化的残基中添加氨基酸(例如酪氨酸)可以减少其对其他氨基酸优化的影响。在第一轮计算之后,可以根据误差输出在两个部分之间切换残差。氨基酸分析结果通常用其组成氨基酸残基的峰高或峰面积表示。如果您需要分析生物学数据,可以下载此aminoXpress软件!
安装方式:
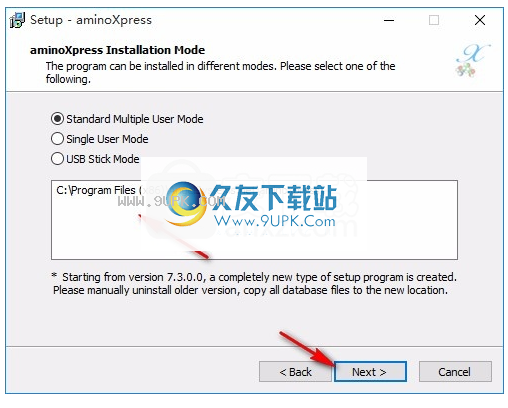
1.打开aminoXpressSetup7700.exe提示安装协议的内容

2,程序可以以不同的方式安装。请选择以下选项之一。
标准多户模式
单用户模式
USB记忆棒模式
3.提示安装软件的界面,该软件安装在C:\ Program Files(x86)\ AngelSystems.net \ aminoXpress \ HelpHTML中
4.提示安装结束,单击“完成”以打开主程序
5.软件界面如图所示,如果可以使用aminoXpress,则启动一个新项目
软件特色:
1.氨基酸分析:此模块计算氨基酸分析结果的最佳残留率。通过将在水解过程中容易氧化的残留物输入“未优化的残留物”部分,可以将对优化结果的影响降至最低。
2.复合积木的组成:该模块基于以下事实:积木(氨基酸,片段)具有离散的权重值以识别意外产物。扫描它们的组合后,最小的偏差更有可能是衍生产品。
3.消化模式:此模块通过用户定义的酶或化学模式预测消化产物。
元素分析:此模块计算序列中不同元素的理论百分比。通过进一步输入每种元素的实验百分比组成,可以确定其经验公式和/或其聚集体。
4.碎裂:此模块预测质谱图中三种键断裂产生的碎裂:一氧化碳/一氧化碳,一氧化碳/碳氢化合物和碳氢化合物/碳氢化合物。可以将生成的碎片直接导入质量模块以模拟质量分布模式。
5.螺旋轮/网:此模块显示并操作螺旋结构的顺序轮/网显示。
6.同位素分布图:该模块预测输入公式或序列的同位素分布图,电荷的或模式从1变为3
7. Lib组合库:此模块以1-3电荷或模式预测组合库(例如肽库)的质谱。直到最后一步才能执行质量舍入,以确保准确性。此外,该模块将其预测能力扩展到有机文库:随机变体可以是氨基酸,片段或两者。结果还包括随机截面残差组合
8.主题发现:此模块使用模式识别来预测序列中潜在的药物活性位点。它有助于扫描显示相同生物学活性的预设模式或随机重复模式,潜在药效基团和加载序列。
9.分子量:此模块计算肽和蛋白质的分子量。输入可以通过常规的序列方案或通过诸如氨基酸和片段的结构单元来完成。替代方案使模块能够计算复杂分子的分子量,例如具有保护基团的肽,拟肽,长分子和支链分子。原子量的进一步使用通常会扩展模块计算任何化合物分子量的能力。
10.数据库数据库管理:该辅助模块允许用户管理该程序中使用的所有数据库,例如氨基酸数据库,片段附件数据库等。开放式设计允许用户根据其特定项目自定义数据库。
11. Sequence Sequence Loader:此辅助模块允许用户将序列加载和解析到相应模块的入口部分。专家系统会自动识别属性,例如侧链,环结构,错误取代和其他加合物。
软件特色:
分子量
该程序通过使用氨基酸,片段和元素作为结构单元来计算化合物的分子量。双向输入协议使该程序能够轻松处理常见和复杂的序列。内置的肽段智能功能可以正确解析大多数序列(环状,分支等)。当序列变得过于复杂时,输入备用残基的协议即会生效。对元素原子量的强烈异常处理使该程序能够计算自然界中任何分子的分子量。
肽/蛋白质序列
p中的大多数建筑图案
肽/蛋白质序列是结构成分,只有某些片段是功能实体,通常称为活性位点或基序。传统主题搜索通常需要输入现有已知模式。从大量序列中提取通用模式是一项繁琐的任务。该模块旨在自动找出那些常见的图案,并根据其频率对其进行排序。它特别适用于分析具有共同生物学功能的一组序列。
以两种模式构建:扫描和模式匹配。扫描模式是扫描一组具有相似生物学功能的序列,但尚未找到通用模式。模式匹配模式是传统的模式匹配,仅适用于已识别出通用模式的模式。
此模块最好与序列加载器模块一起使用,在该模块中,可以导入定界文本文件中的序列,然后进行分析。因此,可以轻松地连接其他数据库中的序列。
官方教程:
示例:氨基酸分析
为了找到以下实验结果的最佳氨基酸比例:

通过氨基酸分析获得的肽样品:
H-Ala-Phe-Ala-Lys-Phe-Tyr-Tyr-Phe-Glu-OH
1.输入要优化的残留物的名称,数量和峰高/面积信息。
2.输入名称,计数和峰高/峰面积信息,以优化残留。

*允许使用空行,因此在第一轮计算之后,您可以根据残基的百分比误差轻松地在两个输入部分之间切换残基。
3.选择一种优化方法。

4.单击执行按钮。
5.样本条目的优化结果如下:
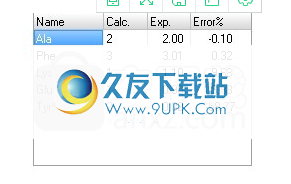
“ *”表示优化中未包含的残基。
码库开关
例
代码1> 3
对于所有输入字段(包括序列),此命令将自动将氨基酸代码从一个字母代码切换为三个字母代码。
代码3> 1
对于所有输入字段(包括序列),此命令将自动将氨基酸从三个字母的代码切换为一个字母的代码。请注意,将三字母代码转换为一字母代码后,某些信息将丢失。即,D氨基酸不能再通过字母代码来识别。
代码库开关
在代码库3下,顶部序列窗格中的血管紧张素I如下:

单击3> 1按钮,顶部序列窗格更改为:

MW模块残渣输入窗格更改为:

如果单击1> 3按钮,反之亦然。
成分组成
例
如果综合产生了与设计不同的意外情况,则此模块将有助于找出其标识。该计算基于以下事实:构件:残基,碎片具有离散的权重值。扫描完它们的组合后,偏差最小的组合很可能是新产品。
目的
输入要排序的分子量,然后输入排序允许的阈值。
筑模块
输入组分的名称,例如氨基酸,片段或元素及其可能的范围:最小。和最大。如果范围不容易确定。然后,您可以输入范围:从“ 0”到将目标分子量除以相应的结构单位后的结果四舍五入。然后,程序将对该构建基块执行全范围搜索。允许为负范围。这种设计的目的是失去一些结构单元,主要是片段,而不是增加分子结构形成副产物。
有时,您可以故意增加Max的值。 。因此,也可以搜索高于理论值的峰。
圆?
对于环状分子,请检查
您怀疑在合成过程中形成的桥:酰胺或二硫键及其最大数目。对于未包括的其他类型的桥梁,可以在“ Building Blocks”部分的负范围内输入在桥梁形成过程中丢失的碎片。也就是说,从-2到0。
比赛名单
结构单元的输出和输入分子量阈值内的分子量的组合。当序列变得复杂时,搜索结果通常包含多个匹配项,特别是如果将阈值设置得很高,则用户将确定每个匹配项的合理性。
方法
选择要用于排序的方法:
组合扫描:用尽所有可能的组合以获得给定范围,并检查其是否匹配。
蒙特卡洛(Monte Carlo):随机生成给定数量的随机样本,并检查它们是否匹配。
建议使用组合扫描,除非您尝试分类的组合数量太大而无法在合理的时间内执行。
执行
开始计算。
明确
清除所有条目和输出。
停
终止计算。
构建基块组成
范例1。
靶肽的副产物:
H-Ala-Ser-Phe-Tyr-Gly-Glu-Lys-Lys-OH
实测分子量:618.7。
要查找此副产品的组成部分的可能组合,请执行以下操作:
1.输入目标分子量和容许误差阈值。

2.在名称及其可能的范围内输入构造块:最小。和最大。

3.如果怀疑发生环化,请输入桥类型:酰胺或二硫键及其最大数目。

4.检查方法。

5.样品输入的结果是:1Ala1Ser2Lys1Gly1Glu,这是一种丢失的肽,带有残基Phe和Tyr。

数据库管理员
例
该程序为用户提供了管理数据库中存储的所有信息的可用性。您可以从所选数据库中添加,更改或删除项目。可以通过浏览数据库并双击感兴趣的项目来实现项目选择。
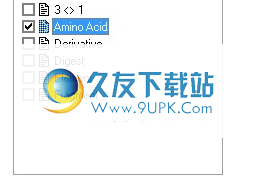
3 <> 1个数据库
数据库包含代码库3和代码库1中的氨基酸缩写。

氨基酸数据库
该数据库包含每种氨基酸的重量信息。

派生数据库
该数据库包含有关派生组的信息。这些是片段或分子,可以导致修饰的产物,或仅导致质量峰移动穿过盐的形成。

抽象数据库
该数据库包含消化中的切割模式。

元素数据库
该数据库在标准周期表中包含原子量信息。

用户应格外注意修改此数据库中的值。如果修饰的目的仅是使用给定元素的同位素之一。最好将同位素添加到“片段附件”数据库中。用于处理两个数据库的算法几乎相同。
作者要对IUPAC表示衷心的感谢,他同意从以下地址(©1994 IUPAC)重新发布标准原子量表:
元素的子量,1993,Pure&Appl。化学,66,2423,1994。
片段数据库***
与氨基酸数据库类似,但有一些例外:附加重量是附加片段的重量与替换片段的重量之差。可能是负面的。公式条目由两部分组成,两部分之间用“ /”分隔,“ /”之前的段本身以及“ /”之后的替换段。即,C端-OH被-NH 2基团取代,分子式如下:NH 2 / OH。如果附加组件是中性分子,例如HCl,则可以省略“ /”。

Ac和NH2是Ac / H和NH2 / OH的缩写,在N端酰化肽或C端酰胺肽中最常见。如果NH2取代了芳环上的氢,则应使用不同的缩写,即NH2_h,并且其公式条目为NH2 / H。
序列数据库
加
将项目(从输入窗格中)添加到相应的数据库。
更改
选择相应的数据库,浏览数据网格,然后双击感兴趣的项目以将其现有信息带到输入窗格中,对其进行修改,然后单击“更改”按钮。由于程序使用第一个字段(即缩写)作为标识字段,因此无法直接更改此字段。要更改身份字段,请携带
现有信息到输入窗格,然后删除该项目。 修改后的信息保留在“输入窗格”中,然后单击“添加”按钮。
另请注意,用户应更新到
指定实体的所有相关项目。例如,要更新氨基酸数据库中的丙氨酸,用户应更新由缩写“ Ala”,“ A”等标识的项目。
删除
选择相应的数据库,浏览数据网格,然后双击所需的项目以在“输入窗格”中显示相关信息,然后单击“删除”按钮。
**对于每个数据库,始终至少保留一个条目以保持其完整性。
***片段数据库特殊说明
·附加重量是附加分段重量和替换分段重量之间的差。可能是负面的。
·片段的公式条目由两部分组成,两部分之间用“ /”分隔,片段附件本身在“ /”之前,而要替换的片段在“ /”之后。即,C端-OH被-NH 2基团取代,分子式如下:NH 2 / OH。如果其他项是中性分子,例如HCl,则可以省略“ /”。
缩写:Ac和NH2保留用于替换Ac / H和NH2 / OH,并且最常见于N末端酰化肽或C末端酰胺肽。如果在其他类型的取代中使用相同的取代基,则片段连接数据库中应使用不同的缩写。例如,如果NH2取代了芳环上的氢,则缩写可能类似于NH2_h,并且公式条目为NH2 / H。
示例:数据库管理器
将该项目添加到数据库
1.选择相应的数据库。
2.单击清除按钮清除输入窗格。
3.输入相关信息。

4.按添加按钮。
更改数据库中的项目
类。非身份字段:
1.选择相应的数据库。

2.在数据库中浏览要更改的项目。双击该项,将原始信息带入“输入窗格”。
3.进行更改,然后按更改按钮。

B.身份字段:
1.选择相应的数据库。

2.在数据库中浏览要更改的项目。双击该项,将原始信息带入“输入窗格”。
3.按Delete按钮将其从数据库中删除。
4.修改保留在输入窗格中的信息(从Bromelainnn中删除“ nn”),然后按“添加”按钮。

从数据库中删除项目
1.选择相应的数据库。

2.浏览数据库中要删除的项目。双击该项目将相关信息带到输入部分。
3.按删除按钮。
4.如果由于刚删除的项目的信息仍在输入窗格中而意外删除了错误的项目,只需单击“添加”按钮将其放回去。

消化
例
顺序
可通过三种方式提交蛋白质或肽段序列进行消化:a)使用序列加载器。 b)直接输入序列。或3)从剪贴板粘贴。
分段
进入序列通过所选切割模式生成的消化产物。
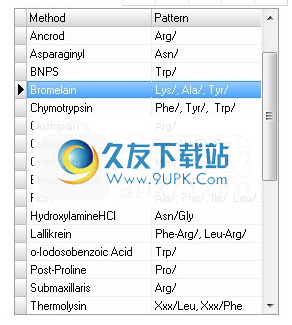
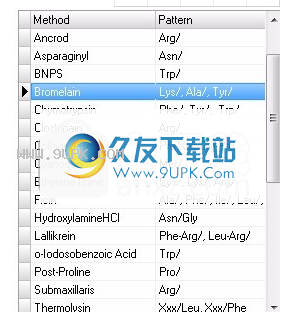
选项
方法选择存储在数据库中的酶或化学裂解方法。
所选方法的破解模式。您也可以直接输入图案。但是,如果您以后希望使用相同的拆分模式。建议您通过数据库管理器将此模式保存为一个名称。使用三个字母代码。一些例子是:
精氨酸/
Arg-XXX / Gly-Glu /
“ /”表示剪切中涉及的键。
准确的分子量分析。
执行
开始计算。
明确
全部清除。
示例:摘要
预测序列的消化产物:
H-Ser-Phe-Leu-Leu-Arg-Asn-Pro-Asn-Asp-Lys-Tyr-Glu-Pro-Phe-OH
1.输入顺序。

2.选择一种消化方法。

3.单击执行按钮开始预测。消化后的产品将在“片段”网格中列出。
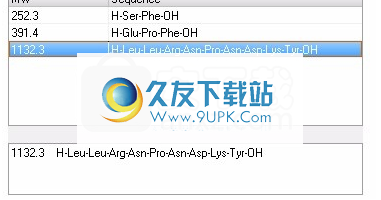
如果剪辑由于显示限制而被截断,您可以双击感兴趣的行以在下面的详细信息字段中显示相应的信息。
元素分析
例
元素分析分为两个步骤:
1)使用Exp。通过输入序列信息作为元素,检查是否未选择合成。 ,然后按Exec按钮,组成元素将显示在Exp中。组成一个网格。理论百分比将显示在结果部分。
2)有经验。检查组合
激活预测模式后,进一步输入实验百分比读数,然后按执行按钮,可能的分子式将显示在结果部分。
元件
输入符号和相应的元素计数。或使用序列加载器功能。单击Seq向下箭头按钮时。目标序列的元素将被解析。如果涉及循环序列,请仔细检查状态栏以获取“分析信息”。在大多数情况下,解析器将正确识别桥类型,酰胺键和二硫键。仅对于某些氨基酸的头尾桥,存在潜在的歧义,可能是酰胺或二硫键。
经验组成
激活后,输入每个元素的实验百分比组成。
“目标FW”例如,如果目标配方重量已知,请输入伴随质谱的值。否则,输入“ 0”。
团聚有时,几种肽的基质会捕获一个或几个小分子,即三个阿片样物质会捕获两个水分子。几种肽也可以形成二聚体或多聚体。该值是扫描范围内聚集的肽的最大数量。
阈值%允许公式权重偏差以进行匹配过滤。如果仅涉及小的添加剂分子(例如TFA,H2O),请设置一个较低的值。如果预期会出现潜在的删除或添加序列,请设置一个较高的值。
执行
开始计算。
明确
清除所有条目和输出。
停
终止计算。
结果
Calc模式:Exp。时间。成分复选框未激活,并且将显示每个元素的理论百分比。
描模式:Exp。组件复选框被激活,程序处于预测模式。除了目标配方中每种元素的组成百分比外,还列出了与实验组成匹配的预测配方。还提供了每个预测公式与相应目标公式之间的差异。
示例:元素分析
要分析肽的元素分析结果:
H-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-OH
哪里

1.输入元素的符号和相应的计数。

2.如果残留物是通过Sequence Loader导入的,请检查状态栏中的“分析信息”以查看是否为循环?信息已正确转换。
3.单击执行按钮。每个元素理论组成百分比将显示在“结果”部分中。将此结果与观察到的实验数据进行比较。如果它们不能很好地匹配,请转到下一步。

4.通过检查Exp激活扫描模式。组成。

5.输入实验结果。

和扫描参数

6.再次单击执行按钮。匹配公式如下:

结果列表中上面突出显示的项目构成2个分子的替换,其中H2O丢失,这可能表明形成了二聚体。