HyperChem是一款专业的化学分子绘制软件。化学分子绘制工具中的佼佼者HyperChem。软件建立具有超湿度的分子非常简单。只需从元素周期表中选择一个元素,然后单击并拖动鼠标以绘制结构草图,鼠标围绕键旋转,立体化学和键的橡皮筋控制使变化结构更容易;提供广泛的选择,突出显示和显示,使用户能够轻松地专注于对复杂分子感兴趣的面积;可以使用方便的鼠标控制工具,旋转,转换和调整结构尺寸。修改控制工具的操作,使用系统中的模型构建器将粗略草图转换为3D结构,可以用各种取代基更换任何选定的氢气(包括您自己的定制更换);易于应用Builder约束:指定关键长度,键角,扭转角度或选定原子的关键几何形状;指定原子型,原子电荷,形成电荷和原子质量,建立簇和复杂的分子组装;移动组等移动单个原子和分子如此简单;肽和核酸,突变残留物由氨基酸和核苷酸残余文本经销制成,逐渐构建大分子,其可以添加用于水救援的周期性均衡水分子,并且可以使用周期性边界条件。在其他溶剂系统中,也可以使用溶剂;用户需要下载体验。
软件功能:
从标准文件格式导入结构:Brookhaven PDB,ChemDraw ChM,MOPAC Z-Matrix,MDL Mol和Tripos Mol2文件。
分子显示
用球和杆,融合的CPK球,蝙蝠,球和气缸或管道显示器结构
在任何渲染中添加范多瓦点。
在同一分子中的任何原子上使用任何渲染。
指定杆或圆柱的宽度,以及球的半径。
立体声和透视和质量设置可以使用。
分子的光跟踪图像显示在工作空间中。
选择并命名原子集以自定义显示或监视属性。
设置并显示原子的自定义标签。
显示键标签,显示当前关键长度或电流计算的量子机械密钥顺序。
使用带,β折叠,随机线圈,圆柱等和可选的侧链显示蛋白质骨架。
突出潜在的氢键相互作用。
娃娃扭矩矢量和梯度矢量。
计算化学
通过单一,几何优化或转换状态搜索计算,使用超级探索量子或经典模型潜在能量。
包括热运动和分子动力学,兰西林动力学或大都会蒙特蒙特仿真。
您可以添加用户定义的结构约束。
计算类型
单点计算可以确定给定固定几何形状的分子能和性质。
几何优化计算使用能量最小化算法定位稳定性结构。提供五种最小化算法。
振动频率计算可以找到优化结构的正常振动模式
可以显示振动谱,并且可以为特定转变相关联的振动运动。
转换状态搜索使用特征向量来遵循或同步与过渡状态对应的转移结构,然后计算分子特性。
分子动力学模拟分子系统的经典轨迹,并且量子力可用于模拟反应碰撞。
加热,平衡和冷却时间可用于模拟与温度相关的退火和其他相关过程。
可以使用恒定能量和恒定温度模拟。
Langevin Dynamics仿真对常规分子动力学增加了摩擦和随机力
在不包括显式溶剂分子的情况下建模溶剂碰撞效果。
所有Monte Carlo在给定温度下模拟了统计收集的采样配置
它可用于探索系统的可能配置,并计算温度的平衡平均值。
由单励磁配置交互(CI)产生的激发状态。
安装步骤:
1.用户可以单击本网站上提供的下载路径以下载相应的程序安装包。
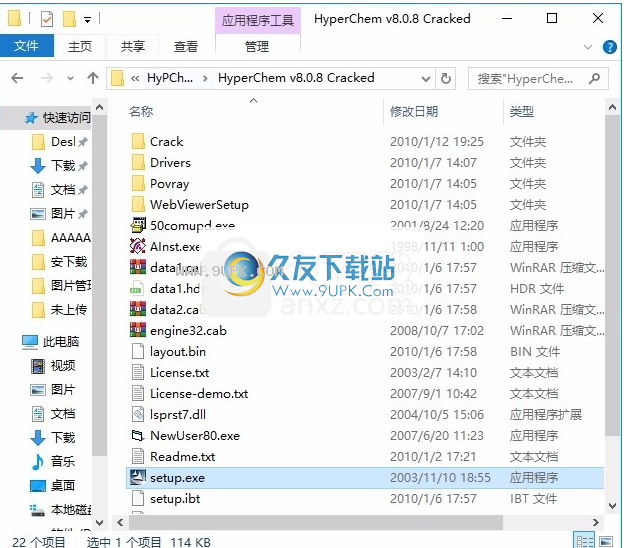
2,只需要使用解压缩函数打开压缩包,双击主程序安装,弹出程序安装界面
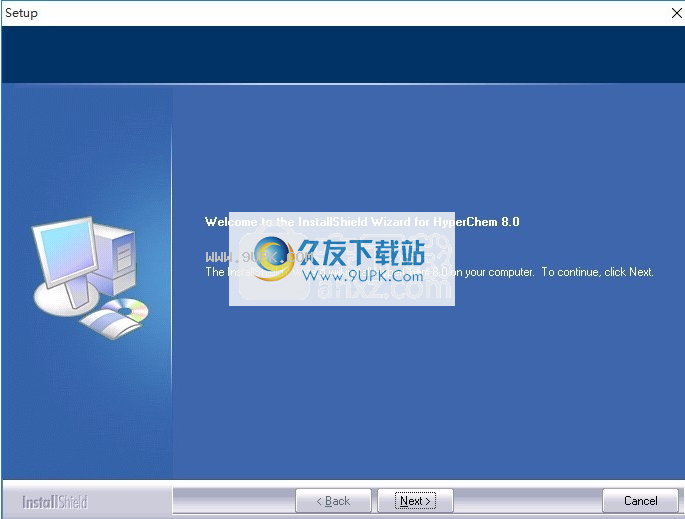
3,同意上述协议条款,然后继续安装应用程序,单击同意按钮
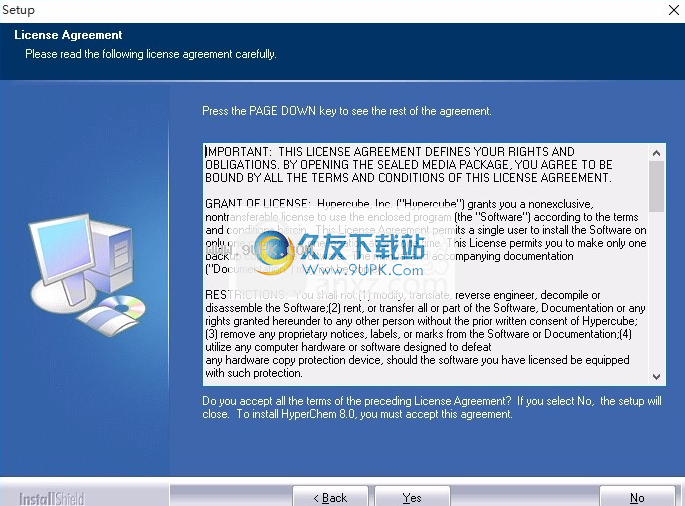
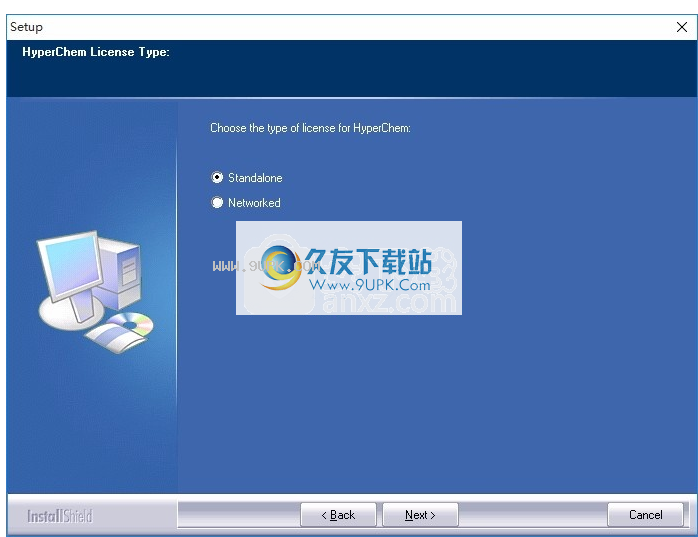
4,弹出以下界面,用户可以使用鼠标直接单击下一个按钮

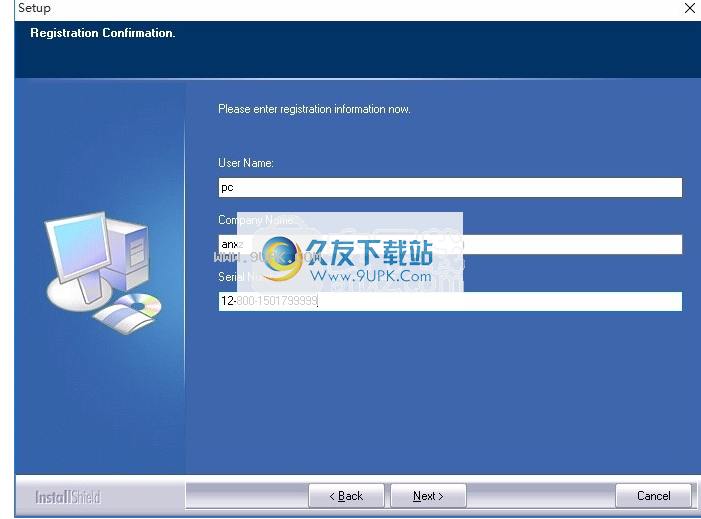
5,弹出激活码输入接口,输入ANXZ,激活码输入:12-800-1501799999
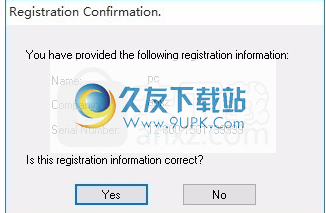
6,然后弹出提示,输入程序序列号:12-800-150179999
7,您可以根据自己的选择单击“浏览”按钮,您需要更改应用程序的安装路径。
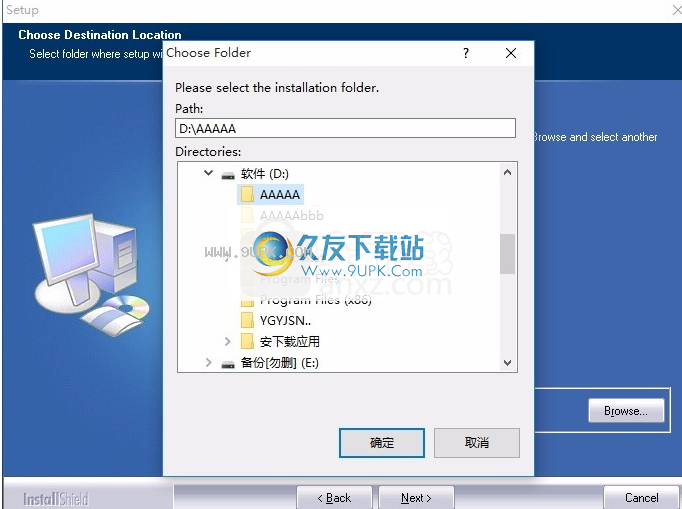
8,弹出以下界面,用户可以使用鼠标单击下一个按钮,可以根据您的需要安装不同的组件

9,弹出应用程序安装计划加载界面,只等待负载完成
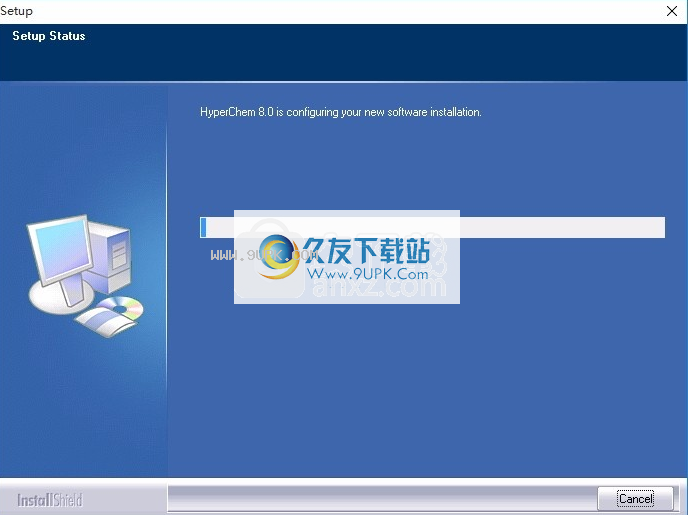
10.单击根据提示的安装,弹出程序安装完成界面,单击已完成的按钮

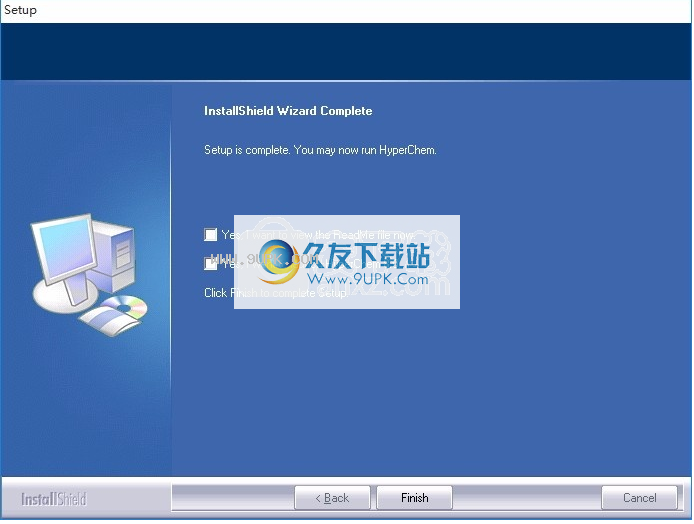
裂缝方法:
1.安装程序后,不运行程序,打开安装包,然后将文件夹中的裂缝文件复制到粘贴板。
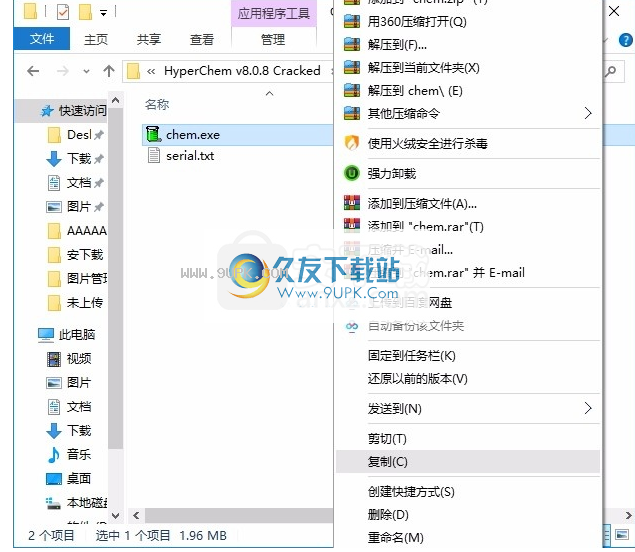
2,然后打开程序安装路径,将复制的裂缝文件粘贴到相应的程序文件夹替换源文件(程序文件夹的路径)
小编更改文件路径为D:\ AAAA \程序
3.完成上述步骤后,您可以双击应用程序以打开它。您可以获得相应的裂缝计划。

使用说明:
该版本中的新功能包括配置交互式计算功能,除了通常的单一感应CI,还有双激发CI(DCI)或双引起单个CI(SDCI)的功能。此新功能仅适用于AB Initio Calculation。它只使用主内存完成,因此非常大的DCI计算可能不是实用的。
此外,核酸(Perez等人,生物物理轴颈,92,3817-29)的新参数可用于分子机械琥珀琥珀琥珀·琥珀色,Amber96和Amber99参数。这些新值是在传统的
DNA或RNA连接中的原子型CT被新的原子CI替换。
在创建双站立DNA时,Windows 7和Vista而不是XP错误是在8.0.10中修复的.Windows 7和Vistas在8.0.10中修复。
主要的新功能增加了重要的新帮助。其他帮助引擎对于描述菜单,对话框最有用。此外,已知在最新版本中得到解决。
NAP2Text实用程序(仅限Windows)已更新以对应于Windows 8.0.9及更高版本的超象。此实用程序将分子动态快照文件从二进制文件转换为文本。由于超多年来的超级数据结构发生了变化,因此它只能确保该实用程序将读取当前快照文件和超象未来版本的Windows的快照文件。
您现在可以免费获得8.0.9版本的Windows的超象。
此版本的主要新功能是它可以作为iPhone,iPod和iPad个人服务器(PS)。网络对话框已得到增强,可以显示要使用的当前IP,并允许PS用户输入登录ID和密码以供移动设备使用。此外,日志允许PS用户限制移动设备可以执行的脚本。这允许PS用户为使用移动设备的学生设置教育实验。
Mobile HyperChem免费允许您在设备上绘制,创建和保存分子,并使用一般的超象工具操作,例如“选择”,“旋转”,“翻译”。
此外,移动超频级别1可以与Internet上的Windows复制的超象交互,以返回分子,执行计算并检索结果以在移动设备上显示。 Windows HyperChem充当移动设备的个人服务器(PS)。从某种意义上说,这是一个PS,它只能与设备交互并登录服务器(超冲突)。可用作PS的超象版本必须是超级仓检8.0.9或更高版本。
HyperChem使用OpenGL图形,并使用良好的驾驶员效果良好的显卡。
此支持下,许多标准的Linux问题都很薄弱。要使超胸(或甚至满意)使用其OpenGL图形,您可能需要安装特定的图形驱动程序。
Fedora Core
例如,Fedora Core 10或11开发的超勾拳通常需要下载图形驱动程序。大多数标准图形卡来自NVIDIA或ATI。目前,ATI Divers似乎甚至可用于Fedora Core10。安装Fedora Core 10后必须下载NVIDIA卡驱动程序。所需的特定驱动程序取决于特定的NVIDIA卡。
除Fedora Core外,其他版本的Linux还可以提供适当的图形驱动程序,但不能保证这一点,因此您可能需要搜索Web上的相应驱动程序。 Hyperchem应该与任何驱动程序运行,但除非驾驶员和相关卡支持比最低入境级别更好的图形,否则可能无法正常运行。
HyperChem需要支持其OpenGL渲染图形驱动程序,越来越好,新的Linux发行版可能满足超象的需求,或者至少使其变得简单。
图书馆
HyperChem需要许多库,这是在此阶段的动态库,而不是静态库。动态库(* .so)必须是Linux版本的一部分。这些库可能不是Linux版本的标准安装的一部分,您可以稍后安装它们。最好在安装Linux时添加各种编程工具,以便在一定程度上赋予感觉,成为Linux计算机的一部分。
软件特色:
计算方法
密度函数理论(DFT)
从各种交流和关联功能中选择。
从各种集成网格中选择
从业务中计算和基本功能的所有选项(见下文)。
丰富量子力学
从许多常用的基础(STO-1G到D95 **)中选择,包括标准STO-3G,3-21G,6-31G *和6-31G **基集
可以将其他基本功能(S,P,D,SP,SPD)添加到单个原子或原子组。
用户还可以轻松定义自己的基础组或使用记录的基本集文件格式修改现有基集
Y Hyperchem。
半体验
ħperchem提供11个半经验分子轨道,采用有机和初级化合物选项,过渡金属配合物和光谱仿真。
从扩展的哈奇克,CNDO,Indo,Mindo / 3,MNDO,MNDO / D,AM1,RM1,PM3,Zindo / 1和Zindo / s。
分子力学
四个力场提供了一种计算分子系统探索的稳定性和动力学的方法。
用户定义的原子类型和参数增加了灵活性。
从m(通用领域)和三个专业的生物分子电源领域选择:琥珀色,生物和opls。
混合模式计算
Hyperchem允许您对分子系统的一部分(例如,溶质)进行量子计算,而系统的其余部分被处理。
该边界技术可用于所有量子方法,并与扩展有一些限制。
使用化学家的开发人员套件定制和扩展超杂物
简化超级冲突菜单。添加新图形和计算功能;为特定应用程序创建自定义菜单。
与Visual Basic,C,C和Fortran程序的接口。添加对话框和菜单项。
例如,您可以使用HyperChem来可视化非图形量子化学程序的结构和结果。
超级冲突过程链接到其他Windows程序,例如MS Word和Excel;
将所选结果指向这些应用程序以促进分析和报告。















![小学数学练习机下载37.0中文免安装版[数学学习软件]](http://pic.9upk.com/soft/UploadPic/2012-1/201212417564265012.gif)
![口算练习器 1.0免安装版[算术题练习软件]](http://pic.9upk.com/soft/UploadPic/2013-12/2013123114174412954.jpg)